cystic fibrosis pancreatic insufficiency
 Alcohol Affects a Gene Related to Cystic Fibrosis Promoting the Development of Pancreatitis
Alcohol Affects a Gene Related to Cystic Fibrosis Promoting the Development of PancreatitisLearn about cystic fibrosis, a genetic disorder that affects the lungs, pancreas, and other organs, and how to treat and live with this chronic disease. CF is a rare genetic disease found in some 30,000 people in the USA. If you have CF or are considering testing it, knowing about the role of genetics in CF can help you make informed decisions about your health care. If you or your child have just been diagnosed with cystic fibrosis, or your doctor has recommended tests for FQ, you may have many questions. Diagnosing the FQ is a multi-step process. A complete diagnostic evaluation should include a newly-born screening, a sweat chloride test, a genetic or carrier test, and a clinical evaluation at a care center accredited by the CF Foundation. The increase of a child with cystic fibrosis can raise many questions because the CF affects many aspects of your child's life. Here you will find resources to help you manage your child's daily needs and find the best possible CF care. Living with cystic fibrosis comes with many challenges, including doctors, social and financial. By learning more about how you can manage your disease every day, you can help find a balance between your busy lifestyle and your CF care. People with IC live longer and healthier lives than ever. As an adult with CF, you can reach key milestones that you might not have considered. Planning for these life events requires careful thinking while making decisions that can affect your life. People with cystic fibrosis live longer and more satisfactory lives, thanks in part to the specialized care of CF and a variety of treatment options. The care centers accredited by the Fundación Cistic Fibrosis provide specialized care and management of specialized diseases to people living with cystic fibrosis. We provide funding and credit more than 120 care centres and 53 affiliated programmes at the national level. The high quality of specialized care available throughout the network of the care center has led to improving the length and quality of life of people with FQ. The Cystic Fibrosis Foundation provides standard care guidelines based on the latest research, medical tests and expert consultation on best practices. As a doctor, you are critical to helping people with CF to maintain their quality of life. We pledge to help you partner with patients and their families by providing resources that you can use to improve and continue to provide high quality care. As part of the Cystic Fibrosis Foundation mission to help improve the lives of people living with cystic fibrosis, the PSDC initiative takes advantage of the CF community to report on key efforts to support the management of daily care. Your cystic fibrosis care team includes a group of CF health professionals who associate with you to provide specialized and comprehensive CF care. Many people living with cystic fibrosis and their families face complicated care-related problems they need. Cystic Fibrosis Foundation Compass ensures that no one has to do it alone. CF Foundation Compass is a service that helps people with CF and their families navigate insurance options, connect to legal information and experts, find available financial resources and address other life problems. The members of the CF care team are paramount in providing highly specialized care to people living with CF. CF Foundation Compas can help by serving as a strategic ally for care teams, so team members can focus on caring for their patients. CF Foundation Compass can help you navigate safe, financial, legal, and other problems you are facing. Use this online form to start your conversation with a Compass case manager today. The Cystic Fibrosis Foundation is the world leader in the search for a cure for the CF and supports a wide range of research initiatives to address the disease from all angles. The CF Foundation offers a number of resources to learn about clinical trials and treatments that are being developed to improve the treatment of cystic fibrosis. Our understanding of CF continues to evolve while scientists study what causes the disease and how it affects the body. These ideas drive the development of new and better treatments and bring us one more step to a cure. Researchers, supported by the CF Foundation, have made tremendous progress in improving the health and quality of life of people with FQ. We are committed to providing the tools and resources you need to build continuously on this work. Pancreatic Enzymes Clinical Care Guidelines Pancreatic insufficiency (PI) remains a major problem for most individuals with cystic fibrosis. Recommendations include doses of treatment of substitution of pancreatic enzymes (PERT) in infants, children and adolescents. ShareDownloadsPancreatic Guidelines for Clinical Care Enzymes: Executive SummaryBorowitz DS, Grant RJ Durie PR, the Consensus Committee. J Pediatr. 1995; 127:681-84. Pancreatic insufficiency (PI) remains an important issue for most individuals with cystic fibrosis. Left without treatment, it could lead to growth failure, weight loss, abdominal swelling, scents of odors or diarrhea. The use of pancreatic enzyme replacement therapy (PERT) has helped relieve these symptoms. However, the Cistic Fibrosis Foundation received reports of 35 cases of strict colonization between January 1990 and December 1994, which were considered to be related to high doses of PERT. Guidelines for the dosage of PERT and the avoidance of fibrous colonopathy were published in 1995 based on a consensus conference organized by the CF Foundation together with the United States Food and Drug Administration (FDA). The recommendations included PERT doses in infants, children and adolescents, and a warning to use caution when PERT doses exceeded 2,500 lipase/kg/meal units. Since the publication of the original guideline in 1995, a number of new publications related to the use of PERT in the FF have been published, some of which are mentioned below. Recommendations Most individuals with FQ are insufficient pancreatics. Therefore, the provision of safe and effective replacement of the pancreatic enzyme is a key therapy in the CF. Pancreatic Enzyme Replacement Therapy Recommendations Recommendations Test assessment Insufficient pancreatic patients should eat a high calorie diet with unrestricted fat that is appropriate for age and clinical status. These diets have been higher than low-fat diets to promote growth and lung function. Consensus A nutritional assessment should be done regularly as a routine care component in patients with CCI. An additional evaluation should be performed when the dosing of PERT is altered. Consensus The dose of enzyme can be done either by grams of ingested fat or by weight. The fatty dose is more likely to measure the normal pancreatic response to a meal, although the weight-based dose can be simpler and more convenient, especially in older children and adults. Consensus Babies usually require 450-900 units of lipase/g of fat, OR 2,000-4,000 units of lipase for 120 ml of formula or when they breastfeed. Babies usually ingest a greater amount of fat/kg body weight than adults. Consensus Older children and adults usually require 500-4,000 lipasa units per gram of ingested fat (medium, 1,800 lipasa/g of fat), OR 500-2,500 units of lipasa/kg/meal, 250-1,250 units of lipasa/kg/snack, with three meals and two to three snacks per day. It is suggested that the initial dosage is in the lower range and translated as necessary to treat malabsorption. Consensus The doses of enzymes above 2,500 lipasa/kg/meal units, or 4,000 lipasa/g units of fat justify further research. The doses of enzymes √6,000 units of lipase/kg/meal have been associated with fibrous colonopathy. It is unclear whether the doses ny 2,500 units of lipase/kg/meal or ny4,000 units of lipase/g of fat are safe. It is also unlikely that the doses of higher enzymes will improve the clinical condition in patients with IC and poor growth or gastrointestinal symptoms, so it is recommended not to exceed these doses and that patients with higher dosages are titrate down to a lower dosage range. Consensus Patients should receive only the product brands prescribed by their CF care center. Micro-encapsulated enzymes with ventricular encoding are the most effective treatment for IP in the CF. Patients should not use enzymes or health enzymes in the food store without an entrum coating unless it is indicated to do so by their CF doctor. Consensus Enzymatic capsules can be opened and the content mixed with a small amount of apple puré or other non-alkaline food, but should not be crushed or allowed to sit in the food. Enzymes can be inactivated by exposure to alkaline environments or prolonged contact with a wet environment. Consensus Enzymes should be stored in a cool and dry place and checked regularly for expiration dates. Consensus Signs and symptoms of poor response to enzyme therapy include abdominal complaints (blockers, flatus, abdominal pain, and loose feces, frequent or diarrhea in excess) along with symptomatic steatorea (bulky, fat, fetal stools) and/or poor growth. These symptoms can also be seen with other conditions. Before increasing enzymes as a result of symptoms, consider dietary factors, adhesion problems, intestinal hyperacidity, abnormal intestinal motility and liver disease with low salt content of intestinal bile, as well as non-CF gastrointestinal disease. Consensus Fibrous colonopathy should be considered in patients with CCI who have evidence of obstruction, bloody diarrhea, or cylide asciitis, or who have a combination of abdominal pain, continuous diarrhea and/or poor weight gain. Fibrous colonopathy is characterized by strict colonies. Its cause is unclear, but it has been associated with high doses of pancreatic enzyme supplements. Patients with the highest risk include children under 12 years of age, patients with Ø6,000/kg/meal lipase units for more than 6 months, a history of meconium ileum in childhood or distal bowel obstruction syndrome, and history of prior intestinal surgery. Diagnosis is usually performed by image or histopathology. Fibrous colonopathy can respond to the reduction of the dose of enzymes, particularly in early stages, but in later stages it may be required to colectomy. Consensus Insufficient pancreatic patients should eat a high calorie diet with unrestricted fat that is appropriate for age and clinical status. These diets have been higher than low-fat diets to promote growth and lung function. Consensus A nutritional evaluation should be done regularly as a routine care component in patients with CCI. An additional evaluation should be performed when the dosing of PERT is altered. ConsensusThe dose of enzyme can be done either by grams of ingested fat or by weight. The fatty dose is more likely to measure the normal pancreatic response to a meal, although the weight-based dose can be simpler and more convenient, especially in older children and adults. ConsensusInflators usually require 450-900 units of lipase/g of fat, OR 2,000-4,000 units of lipase for 120 ml of formula or when they breastfeed. Babies usually ingest a greater amount of fat/kg body weight than adults. ConsensusChildren and older adults usually require 500-4,000 lipase units per gram of ingested fat (average, 1,800 units of lipase/g of fat), OR 500-2,500 units of lipase/kg/meal, 250-1,250 units of lipase/kg/snack, with three meals and two to three snacks per day. It is suggested that the initial dosage is in the lower range and translated as necessary to treat malabsorption. Consensus The doses of enzymes above 2,500 units of lipase/kg/meal, or 4,000 units of lipase/g of fat justify further research. The doses of enzymes √6,000 units of lipase/kg/meal have been associated with fibrous colonopathy. It is unclear whether the doses ny 2,500 units of lipase/kg/meal or ny4,000 units of lipase/g of fat are safe. It is also unlikely that the doses of higher enzymes will improve the clinical condition in patients with IC and poor growth or gastrointestinal symptoms, so it is recommended not to exceed these doses and that patients with higher dosages are titrate down to a lower dosage range. ConsensusCustomers should receive only the product brands prescribed by their CF care center. Micro-encapsulated enzymes with ventricular encoding are the most effective treatment for IP in the CF. Patients should not use enzymes or health enzymes in the food store without an entrum coating unless instructed to do so by their CF doctor. ConsensusEnzymatic capsules and content mixed with a small amount of apple puré or other non-alkaline food can be opened, but they should not be crushed or allowed to sit in the food. Enzymes can be inactivated by exposure to alkaline environments or prolonged contact with a wet environment. ConsensusNew Questions Answerless QuestionsRead more This executive summary was prepared by:Sarah Jane Schwarzenberg, M.D. (Minnlin Health University) and Jill Dorsey, M.D., M.S. (Nemours Children's Specialty Care) The guidelines were issued in November 1995, revised in April 2019 and no update was determined at this time. . The reference to any specific product, process or service does not necessarily constitute or imply its approval, recommendation or favoration by the Foundation of Cistics Fibrosis. In addition, the emergence of external hyperlinks does not constitute the endorsement of the Foundation for the Cystic Fibrosis of linked websites, nor the information, products or services contained therein. The information contained on this site does not cover all possible uses, actions, precautions, side effects or interactions. This site is not designed as a substitute for the treatment council of a medical professional. Talk to your doctor before making changes to your treatment. The approved drug information is available in . Share this pageRelated TopicsBeyond ReadingFollow us to find an eventWith over 70 chapters and offices across the country, there are many ways to get involved. Cystic Fibrosis Foundation Montgomery Avenue 4550. Suite 1100 N Bethesda, MD 20814301-951-4422 800-344-4823 (cost-free)

Exocrine pancreatic insufficiency causes, symptoms, diagnosis & treatment

Cystic Fibrosis & Nutrition – Digestive Topics

Introduction and practical approach to exocrine pancreatic insufficiency for the practicing clinician - Othman - 2018 - International Journal of Clinical Practice - Wiley Online Library

Frontiers | Cystic Fibrosis of the Pancreas: The Role of CFTR Channel in the Regulation of Intracellular Ca2+ Signaling and Mitochondrial Function in the Exocrine Pancreas | Physiology

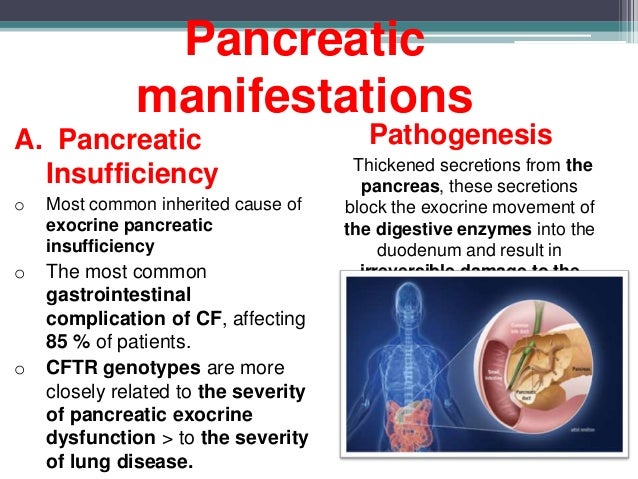
Cystic Fibrosis- Pathophysiology -Managment- TeachMePaediatrics

A Primer on Exocrine Pancreatic Insufficiency, Fat Malabsorption, and Fatty Acid Abnormalities

Cystic Fibrosis | SpringerLink

Introduction to Pancreatic Disease: Chronic Pancreatitis | Pancreapedia

Cystic Fibrosis Challenges Proper Digestion and Nutrient Absorption

Cystic Fibrosis: An Overview of the Past, Present, and the Future - ScienceDirect
Cystic Fibrosis "Overview of Gastrointestinal Diseases"

Nutrients | Free Full-Text | Night Blindness in Cystic Fibrosis: The Key Role of Vitamin A in the Digestive System | HTML

Pancreatitis and pancreatic cystosis in Cystic Fibrosis - ScienceDirect

FDA | Jess Brantner- WVU Dietetic Intern

Chronic pancreatitis and cystic fibrosis | Gut

Gastrointestinal manifestations of cystic fibrosis: A primer for pediatricians

Exocrine pancreatic insufficiency - Wikipedia

Cystic Fibrosis | ThinkPEI

Basics of Pancreatitis and Pancreatic Exocrine Insufficiency
. | Semantic Scholar")
Figure 1 from Common clinical features of CF (respiratory disease and exocrine pancreatic insufficiency). | Semantic Scholar

vitamins | Jess Brantner- WVU Dietetic Intern
/exocrine-pancreatic-insufficiency-4177936-821-618578bea845406fa96c78167aff8657.png "Exocrine Pancreatic Insufficiency: Symptoms, Causes, and Diagnosis")
Exocrine Pancreatic Insufficiency: Symptoms, Causes, and Diagnosis

Cystic fibrosis

What is pancreatic insufficiency?

Model for determining the contribution of genetics and environment to... | Download Scientific Diagram

Cystic fibrosis from the gastroenterologist's perspective | Nature Reviews Gastroenterology & Hepatology
PLOS ONE: Abdominal symptoms in cystic fibrosis and their relation to genotype, history, clinical and laboratory findings

Human pancreatic exocrine response to nutrients in health and disease | Gut

cystic fibrosis STUDENT version
![Who's at Risk of Exocrine Pancreatic Insufficiency? [Infographic] | Everyday Health](https://images.everydayhealth.com/images/digestive-health/exocrine-pancreatic-insufficiency/cs-exocrine-pancreatic-insufficiency-risk-infographic.jpg?sfvrsn=3a107997_0 "Who's at Risk of Exocrine Pancreatic Insufficiency? [Infographic] | Everyday Health")
Who's at Risk of Exocrine Pancreatic Insufficiency? [Infographic] | Everyday Health
, 7th Ed.")
Disorders of the Exocrine Pancreas - Pathophysiology of Disease: An Introduction to Clinical Medicine (Lange Medical Books), 7th Ed.

Exocrine Pancreatic Insufficiency: What It Is and Who's at Risk

Gastrointestinal Complications of Cystic Fibrosis - Clinical Gastroenterology and Hepatology

Exocrine Pancreatic Insufficiency: Seen but Not Recognized?

Structure-Function Relationships of CFTR in Health and Disease: The Pancreas Story | Pancreapedia
 Therapeutics and Diagnostics Market Share Demand, Insights and Forecast up to 2025 — Teletype")
Exocrine Pancreatic Insufficiency (EPI) Therapeutics and Diagnostics Market Share Demand, Insights and Forecast up to 2025 — Teletype

Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis | European Respiratory Society

Cystic Fibrosis | American Academy of Pediatrics

Managing EPI and Cystic Fibrosis | Everyday Health

Less Common Causes of Exocrine Pancreatic Insufficiency Explored - MPR
{kind=link}
Posting Komentar untuk "cystic fibrosis pancreatic insufficiency"